





서 론
현행 「식품의 기준 및 규격」의 「식품 등의 한시적 기준 및 규격 인정 기준」에서는 국내에서 식품으로 섭취경험이 없는 새로운 식품원료에 대해 안전성 등을 평가해 식품공전에 등재될 때까지 신청한 업체에 한해 한시적으로 식품원료로 인정받을 수 있도록 하고 있다(식품위생법 제7조 제2항 및 식품위생법 시행규칙 제5조). 이와 같이 국내 식품으로 섭취경험이 없는 원료에 대해 유럽(European Union, EU)에서는 novel food, 미국 Food and Drug Administration (FDA)에서는 Generally Recognized as Safe (GRAS), 그리고 호주·뉴질랜드 Food Standards Australia New Zealand (FSANZ)에서는 novel food와 같은 제도하 인정하여 관리하고 있다. 국내에서 개발된 새로운 식품원료의 해외 수출과 국외 신기술 적용 개발 식품 및 전통식품 등의 수입 등을 위해서는 각 국가에서 식품원료로 섭취를 위한 인정 절차가 요구되는데, 국가마다 인정 조건 및 절차가 다르므로 본 총설에서는 우리나라 및 주요 국가별 새로운 식품원료의 인정 기준 및 절차에 대해 다루고자 한다.
대한민국
「식품위생법 시행규칙」 제5조 제1항에 따른 식품 등 한시적 기준 및 규격의 인정 대상 제품의 인정 대상 범위는 다음과 같다(MFDS, 2024a). 단, 식품원료로 사용되는 경우에만 해당된다.
-
① 국내에서 새로 원료로 사용하려는 농산물·축산물·수산물 및 미생물 등
-
② 농산물·축산물·수산물·미생물 등으로부터 추출·농축·분리·배양 등의 방법으로 얻은 것으로서 식품으로 사용하려는 원료
-
③ 세포·미생물 배양 등 새로운 기술을 이용하여 얻은 것으로서 식품으로 사용하려는 원료
즉, 국내 섭취경험이 없는 원료로 가공과정을 거치지 않은 원재료 그 자체이거나 또는 가열, 건조 및 분쇄 등의 가공과정을 거친 원료, 「식품의 기준 및 규격」에 고시되어 있는 원료이지만, 사용부위가 다르거나 특정 식품유형에만 제한적으로 사용 가능한 원료를 다른 용도 및 수준으로 사용하고자 하는 경우, 국내 섭취경험이 없는 농·축·수산물 등으로부터 추출·농축·분리·배양 등의 가공과정을 거쳐 제조된 원료 및 기존 사용 가능한 식품원료에 나노기술 등 새로운 기술을 이용하여 생산된 원료가 포함된다. 그러나, 오·남용 우려가 있어 국민 정서상 허용이 어려운 원료(예: 체중조절 목적 등), 원료로부터 추출된 특정 단일성분(예: 지아잔틴, 루테인 등)으로 원재료의 특성이 없어진 원료, 광물질과 이로부터 분리, 추출, 농축 등의 방법으로 얻어진 원료, 그리고 기타 식품첨가물, 기능성, 생약 및 의약품 등의 용도로 사용되는 원료는 신청 대상에서 제외된다.
식품 등 한시적 기준 및 규격을 인정받고자 하는 경우에는 한시적 기준 및 규격 인정 신청서(전자문서로 된 신청서 포함)를 작성하여 식품의약품안전처장에게 신청하여야 하는데, 제출자료에는 다음과 같은 내용이 포함되어야 하며 담당부서는 식품의약품안전평가원 신소재식품과이다(NIFDS, 2023).
언제, 어느 나라에서 어떤 경위로 개발되었는지를 기재하며, 특히, 천연물 원재료의 경우에는 그 기원, 학명, 원산지, 사용부위 등을 구체적으로 기재한다. 국내·외 인정·허가 상황, 사용 기준·규격 등의 관련 내용을 정확히 기재하고 관련 증빙자료를 첨부한다. 국제식품규격위원회(Codex Alimentarius Commission, CAC) 등 국제기구에서 검토 중인 경우에는 안전성 평가 상황, 사용기준 및 규격 등 관련 내용을 조사하여 첨부한다. 국내·외에서 식품 등으로 사용실적이 있는 경우에는 사용 용도, 유통량, 제조회사 및 섭취 실태 등에 대한 자료를 제출하는데, 국내에서 국부적으로 민간 사용 경험 관련 자료도 포함되며, 식품으로의 섭취경험이 중요하므로 가능한 많은 자료를 조사하여 첨부하는 것이 바람직하다.
재배 및 사육방법 등에 대한 자료로 원재료에 따른 이전의 재배 및 사육방법 등의 과정을 작성한다. 제조방법 자료로 원재료에서부터 해당원료의 생산에 이르기까지 전체 제조과정을 알기 쉽도록 단계별로 표준화된 공정 흐름도, 각 단계별 세부적 제조방법(사진을 포함한 구체적 작업 내용 및 조건), 그리고 독성물질 있는 것으로 알려진 경우 독성물질 제거 방법에 대한 자료를 잔류기준, 시험법 및 시험법에 대한 검증 자료 등을 포함하여 제출한다. 제조방법은 구체적으로 기재하며, 수입식품인 경우 제조회사가 발행한 자료를 제출하여야 한다. 제조공정에 사용된 용매, 미생물 및 식품첨가물 등에 대한 자료로 제조공정에 사용된 각종 기구의 명칭과 식품첨가물 등의 명칭과 함량에 대한 자료를 제출하는데, 제조 중 사용된 용매, 미생물, 식품첨가물, 기구, 용기 등 안전성 평가와 관련된 모든 사항에 관하여 상세히 기재하고 「식품의 기준 및 규격」(식품의약품안전처고시) 및 「식품첨가물의 기준 및 규격」(식품의약품안전처고시)에 적합한지 확인할 수 있는 자료를 제출한다.
원료를 특징 지을 수 있는 성상, 물성, 섭취 방법, 용도 및 주요 성분 등에 관한 자료를 제출하여야 한다. 원료의 성상으로 최종 신청원료의 육안적 형태, 색, 냄새, 크기 등에 대한 정보를 사진을 포함하여 제출한다. 성분분석 결과로 일반성분(탄수화물, 회분, 조단백질, 지방 등)의 구성 및 함량, 필요시 아미노산, 지방산, 비타민 및 무기질의 구성 및 함량, 약리적 효과를 포함한 성분이 있다고 알려진 경우, 이에 대한 연구·보고 사례와 약리 주요 성분에 대한 분석결과, 시험법 및 검증 자료를 제출한다. 유해물질 분석결과로 중금속, 잔류농약, 미생물, 곰팡이 독소 등에 대한 분석 결과 등을 제출하는데, 식품 등 시험·검사기관으로 지정된 국내 식품위생검사기관에서 분석한 결과여야 한다. 추가적으로 원재료의 특성에 따라 알칼로이드 성분 함유, 항균성 물질 생산, 항생제 내성 여부에 대한 자료 제출이 필요할 수 있다.
안전성 정보 자료로서 해당 원료 또는 원재료에 함유되어 있는 특정 성분의 독성 유무 등 안전성 관련 정보 자료를 수집하여 제출하는데, 국내·외 학술지에 게재된 내용, 국내·외 정부기관 보고서(식품의약품안전처, 농림축산식품부 등 대한민국 정부기관, 미국 FDA, 유럽(European Food Safety Authority, EFSA), Health Canada, FSANZ 등 국외 정부기관) 또는 국제기구(WHO, FAO, CODEX 위원회 등) 보고서 등 공신력 있는 자료를 제출해야 한다. 인체영향 자료로서 해당 원료의 섭취가 영양성분의 흡수·분포·대사·배출에 미치는 영향을 평가한 자료, 해당 원료에 함유되어 있는 영양성분의 자료 및 국내·외 섭취로 인한 건강 부작용 보고 등 정보를 수집하여 제출한다. 알레르기성 자료로 해당 원료로 인해 알레르기 유발에 대한 국내·외 학술지 등에 보고가 있는지 조사하고, 해당될 경우 관련 연구·보고 사례 및 시험결과(혈청실험 및 피부감작시험 자료 등) 등을 제출한다. 독성시험자료는 우수실험실운영규정(GLP)에 따라 운영된 기관에서 실시하고, 경제협력개발기구(OECD) 독성시험방법(OECD Test Guideline)에 따르거나 이에 준하여 시험한 보고서 형태여야 한다. 독성시험자료로는 단회투여 독성시험(설치류), 3개월 반복투여독성시험(설치류) 및 유전독성시험(복귀돌연변이시험, 염색체이상시험, 소핵시험)을 기본으로 제출하여야 하고, 필요한 경우 생식독성시험, 항원성시험, 면역독성시험, 발암성시험에 관한 자료를 추가적으로 요청할 수 있다. 단, 이와 같은 독성시험자료는 미국 GRAS, 유럽연합 novel food, 호주/뉴질랜드 novel food에 등재된 식품원료 및 Codex에 등재 원료의 경우 제출을 면제받을 수 있다.
신청원료의 사용대상식품 및 사용량을 제안하고, 해당 원료에 대한 연령별 평균 섭취량 및 고 소비군의 극단섭취량을 계산하여 제출하는데, 사용대상식품은 「식품의 기준 및 규격」(식품의약품안전처고시)에 명시되어 있는 식품 유형으로 정해야 하고, 섭취량 평가를 위한 근거자료는 최신 ‘국민건강영양조사(보건복지부)’ 자료 등 한국인에 대한 자료를 우선으로 한다. 설정한 사용량이 연령별 극단섭취량(상위 95%) 포함 상한섭취량을 상회하지 않으면 안전성에 문제가 없는 것으로 판단한다.
한시적 기준 및 규격을 인정받은 후 신청인 정보는 변경할 수 있으나, 인정내용인 원료명, 제조기준 및 기타 인정 요건은 변경 대상에서 제외된다. 즉, 제조방법 등이 변경되는 경우 다른 원료로 간주하므로 새롭게 인정 신청을 통해 검토를 받아야 한다. 또한 인정된 원료는 인정받은 자에게만 효력이 있으므로 동일한 원료이더라도 다른 업체가 해당 원료를 제조·수입하고자 하는 경우에는 다시 인정신청을 하여 검토를 받아야 한다.
현행 「식품의 기준 및 규격」에서는 「식품 등의 한시적 기준 및 규격 인정 기준」(식품의약품안전처 고시 제2016-27호)에 따라 인정된 식품원료는 다음의 어느 하나를 충족하면 「식품의 기준 및 규격」 “식품에 사용할 수 있는 원료”의 목록 또는 “식품에 제한적으로 사용할 수 있는 원료”의 목록에 추가로 등재할 수 있다(식품의약품안전처 고시 제2015-78호, 2015.10.29) (MDFS, 2024b).
-
① 한시적 기준 및 규격을 인정받은 날로부터 3년이 경과한 경우
-
② 한시적 기준 및 규격을 인정받은 자가 3인 이상인 경우
-
③ 한시적 기준 및 규격을 인정받은 자가 등재를 요청하는 경우(다만, 인정받은 자가 2명인 경우 모두 등재를 요청하는 경우)
단, 위의 요건 중 하나를 충족하더라도 실질적 생산, 판매, 수입량 등 섭취실적 자료가 부재할 경우 식품공전 등재가 유예될 수 있다. 한시적 기준 및 규격 인정 원료가 식품공전에 등재되면 한시적 인정의 효력은 상실되며, 담당부서는 식품의약품안전처 식품기준과이다.
유럽연합 Novel Food
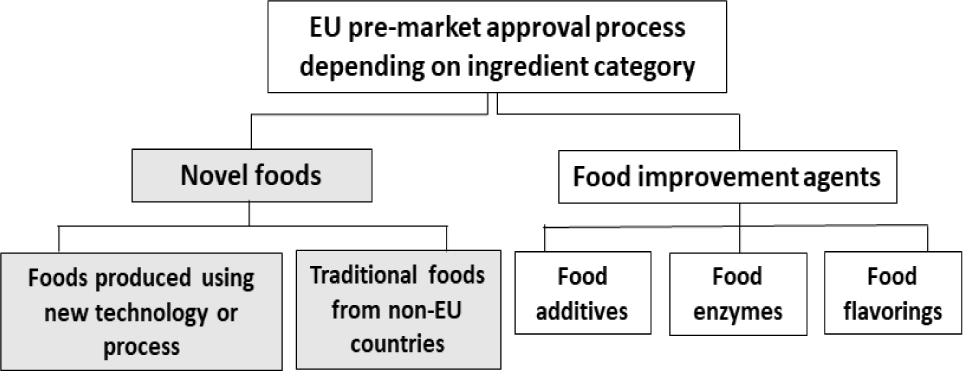
유럽연합에서는 1997년 5월 15일 이전 유럽연합에서 섭취경험이 있는 식품에 대해서는 기존 식품으로 간주하여 유럽연합 일반식품규정 Commission Regulation (EC) No 178/2002에 따라 관리하고 있으며, 시판 전 승인 절차가 필요 없다(European Commission, 2024a: EUR-Lex, 2024a). 한편, 1997년 5월 15일 이전 유럽연합 내에서 광범위하게 섭취되지 않은 식품에 대해서는 Regulation (EU) 2015/2283 규제하 novel food로 분류하고 있고(Fig. 1), 새로 개발된 혁신적 식품, 새로운 기술과 생산공정 사용하여 생산된 식품 및 유럽연합 외 지역의 전통식품을 포함한다(EUR-Lex, 2021). Novel food 목록에서는 사용 조건을 명시하고 있는데, 즉 지정된 식품 범주, 최고 사용량 등을 명시하여 관리하고 있으며, 건조 과일 펄프, 당류, 식품 단백질 및 식물성 오일 등 일부 천연물 기반 식품과 제3국의 전통식품에 대해서는 사용 조건을 특정하지 않고 있다(not specified) (EUR-Lex, 2024b). 유럽연합의 novel food 승인 절차는 다음과 같으며, 유럽연합 규정 2015/2283의 제10조에 명시되어 있다(Fig. 2). 유럽연합 시장에 novel food를 출시하려는 신청자는 해당 법령 및 EFSA의 지침서에 정해진 요건에 따라 유럽연합 집행위원회(European Commission, EC)에 신청서를 제출해야 한다.
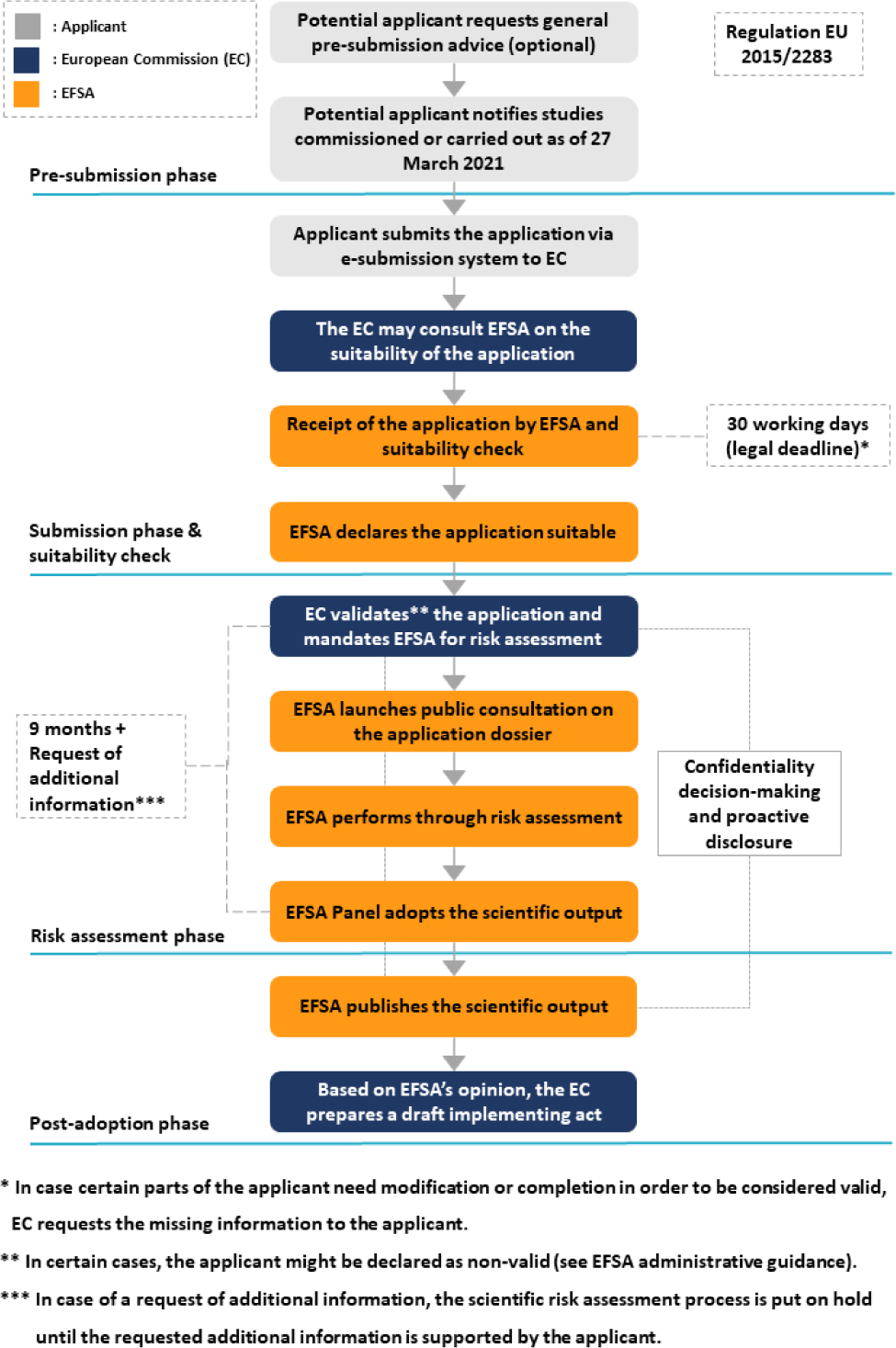
유럽연합의 novel food에서는 유럽연합 이외 지역의 1차 생산물이나 전통 소비 식품에 대해서는 다른 절차가 마련되어 있다(Fig. 3) (EUR-Lex, 2021; EFSA, 2023; EUR-Lex, 2024b). 즉, 이러한 식품은 novel food로 간주되지만 신청서를 통해 승인을 받는 대신 유럽연합 시장에 출시하겠다는 의사 통지서를 제출할 수 있고, 식품의 성분과 최소 25년 동안 최소 하나의 제3국에서 다수의 사람들이 전통 식단에서 지속적으로 섭취한 경험에 대한 자료를 기반으로 안전성을 입증하여 시장에 출시할 수 있다(EU Regulation 2015/2283) (EUR-Lex, 2021). 제3국의 전통식품의 경우, 유럽연합 규정 2015/2283 제14조에 따라 EC에 신청서를 제출하는 간소화된 절차를 진행하고 있어, 평균 5개월이라는 짧은 안전성 평가기간을 가진다. 단, 회원국 또는 EFSA가 안전성에 합리적 이의를 제기할 경우, 유럽연합 규정 2015/2283의 제16조에 따라 안전성 이의 제기와 관련된 문서화된 데이터를 추가하여 제출해야 하며, 법령 및 EFSA의 지침에 정해진 요건에 따라 안전성 평가를 진행해야 한다. 즉, 일반적 novel food 승인 절차와 동일하게 EFSA에서 공개자문 및 위해 평가를 진행한다(EUR-Lex, 2021; EFSA Panel on NDA et al., 2024).

유럽연합에서 일반적인 novel food 승인의 경우와 제3국 전통식품으로의 승인을 위한 안전성 관련 요구되는 데이터는 달라지게 되는데, 일반적 novel food 승인을 위해서는 안전성을 뒷받침하는 광범위한 데이터와 독성학적 연구가 포함되어야 한다. 반면, 제3국의 전통식품에 대해서는 제3국에서의 전통식품을 소비한 섭취경험 및 이력 의한 안전성 평가에 초점을 두고 있다. 현재 총 9종의 제3국 전통식품이 novel food 리스트에 포함되어 있으며, 이들 식품에 대해서는 사용조건이나 사용량을 제한하고 있지 않다(not specified) (EUR-Lex, 2021; European Commission, 2024b; EUR-Lex, 2024b).
유럽연합 novel food에서는 2015년 개정을 통해 최초 승인받은 업체에 한해 요청하는 경우 유럽연합 시장에서 5년간 독점 판매권을 보유할 수 있도록 하고 있다(단, 최초 승인 업체가 판매권을 다른 회사에 양도한 경우 또는 다른 회사가 새로운 과학적 자료를 기반으로 novel food로 승인을 받은 경우 제외). 즉, 새로 개발된 과학적 증거와 독점 데이터는 novel food로 승인된 후 5년 동안 후속 신청자의 이익을 위해 사용할 수 없다(‘data protection’, EU Regulation 2015/2283) (EUR-Lex, 2021).
미국 GRAS
미국 FDA의 GRAS는 Generally Recognized as Safe의 약어로, 연방 식품, 의약품, 화장품법(Federal Food, Drug, and Cosmetic Act)의 201(s) 및 409조에 따르면, 의도적으로 식품에 첨가되는 모든 물질은 시장 출시 전 FDA의 검토와 승인을 받아야 하는 식품첨가물로 간주된다. 전문가들에 의해 해당 물질이 사용 조건에서 안전하다고 일반적으로 간주되거나, 또는 그 물질의 사용이 식품첨가물의 정의에서 예외로 인정되는 경우에는 승인이 필요하지 않을 수 있다(FDA, 2024a).
미국 FDA에서 식품에 새로운 용도로 의도적으로 사용되는 물질에 대한 승인 절차는 Fig. 4와 같으며, 식품첨가물 또는 GRAS로 사용되는지에 따라 다른 절차를 시행하고 있다. 즉, 식품첨가물 정의에 GRAS 재료는 포함되지 않으며, 식품첨가물로 사용을 위해서는 사용 전 FDA의 승인을 반드시 받아야 하는데, 안전성과 관련된 과학적 자료를 제시해야 한다. FDA에서 식품첨가물 또는 GRAS 재료로 평가하여 안전성이 확인된 후에는 식품첨가물 또는 GRAS 재료에 대한 규제 상태는 변하지 않고 그대로 유지된다(FDA, 2024b).

반면, GRAS 재료로 인정(GRAS notice)을 받으면 시장 출시 전 FDA의 검토가 필요하지 않으며, GRAS 물질 인정은 과학적인 절차, 즉 식품첨가물 인정 시와 동일한 양적 및 질적 과학적 자료에 기반하여 또는 1958년 이전 식품으로 섭취된 물질의 경우 일반적 섭취경험에 의해 인정받을 수 있다. GRAS로 인정받기 위해서는 과학적 자료와 물질 사용에 대한 정보가 널리 알려져 있어야 하며, 관련 전문가들 사이에서 해당 물질이 의도된 사용 조건에서 안전하다는 합의가 있어야 한다. GRAS 인정을 위해서는 물질 정보, 제조방법, 사양, 물리적 또는 기술적 효과, 식이 노출량(물질 농도 및 식품 소비량을 고려하여 추정), 자체 사용 제한량, 1958년 이전 일반적 식품 사용 이력(해당되는 경우), 모든 식이 원천과 식단에서 화학적 또는 약리학적 물질을 포함하여 사용 조건에서 일반적으로 안전한 것으로 간주할 수 있음을 입증하는 과학적 자료 및 정보 등을 포함한 설명자료, 그리고 GRAS notice를 뒷받침하는 모든 자료와 정보를 제출해야 한다(CFR, 2024a).
-
1958년 1월 1일 이전에 식품에서 일반적으로 사용된 섭취경험을 바탕으로 안전성이 확인된 경우, 식품첨가물 승인 시에 필요한 것과 동등한 양적 및 질적 과학적 절차가 요구되지 않을 수 있다.
-
1958년 1월 1일 이전에 식품에서 사용된 물질은 미국 이외의 지역에서 섭취경험이 이루어졌더라도 의도된 사용 조건에서 안전하다는 정보를 제공하면 GRAS로 인정될 수 있는데, 식품에서의 일반적 사용은 공개된 정보에 의해 문서화되어 있어야 하며, 해당 물질의 사용 이력과 상황을 확인할 수 있는 독립적 다른 출처의 정보에 의해 확인되어야 한다(최소 2개 이상의 정보 필요).
-
1958년 1월 1일 이전에 미국에서 영양학적 특성으로 널리 소비된 천연물 기원 식품성분은 유해한 영향이 알려지지 않고 안전성에 문제없는 일반적 가공방식으로 처리되었다면 GRAS로 인정될 수 있다.
한편, 공식적인 GRAS notice와 대조적으로 자체 인정 GRAS (self-affirmed GRAS) 절차에 의해 GRAS 재료로 인정받을 수 있는데, 이 과정에서는 기업 또는 개인이 과학적 전문가 패널의 결론을 바탕으로 자사 제품의 안전성을 스스로 선언할 수 있도록 허용한다. 이 경로는 제조업체의 자율성을 제공하지만, 동시에 전문가 패널의 검토를 기반으로 안전성을 보장할 책임이 제조업체에게 있다. 공식적 GRAS notice와 달리 이 경로의 핵심적 특징은 도출된 결과와 결론에 대해 FDA의 검토 절차가 필수적이지 않고, GRAS 결론을 FDA에 통지해야 할 의무도 없으나, FDA의 최종 규칙에서 정한 기준을 충족해야 하며, 안전성에 문제가 제기될 경우 FDA가 자체 인정 GRAS 결정에 이의를 제기할 권한을 보유하고 있다(FDA, 2016; FDA, 2024a, c).
GRAS로 인정된 모든 물질은 현행 우수 제조 관리 기준(good manufacturing practice)에 따라 사용되어야 한다. 즉, 식품에 직접 사용되는 성분은 식품 등급에 적합해야 하고, 식품 성분으로 제조 및 취급되어야 하며, 식품에 첨가된 양이 식품에서 의도된 물리적, 영양적 또는 기타 기술적 효과에 합리적으로 필요한 양을 초과하지 않아야 한다는 요구 사항이 포함된다(CFR, 2024b). 사용 조건에 대한 제한 없이 GRAS로 인정된 경우가 있는 반면, 제한된 사용 조건에서 성분의 안전성을 평가받은 경우 식품의 사용 범위, 기능적 용도 및 사용 수준 등을 포함하여 해당 제한된 범위 내에서만 식품으로 사용되어야 한다(CFR, 2024b).
호주·뉴질랜드 FSANZ의 Novel Food
1991년부터 FSANZ는 호주·뉴질랜드에서 섭취경험이 없는 비전통식품으로 식품 공급 전 안전성이 확보되어야 하는 식품을 non-traditional food, novel food로 간주하여 Australia New Zealand Food Standards Code, Standards 1.1.1 and 1.5.1에 따라 규제하고 있으며, 사용량, 사용 제한 및 표기 요구사항을 포함한 사용 조건을 명시하고 있다. FSANZ에서 정의하고 있는 비전통식품(non-traditional food)이란 다음과 같다(Australian Government, 2024).
-
① 호주 또는 뉴질랜드에서 섭취이력이 없는 식품
-
② 식품에서 유래한 물질로서 호주 또는 뉴질랜드에서 해당 식품의 구성성분 이외의 형태로 섭취 이력이 없는 물질
-
③ 호주 또는 뉴질랜드에서 식품으로 섭취 이력이 없는 물질이나 그 원료에서 유래한 기타 물질
Novel food 여부는 Advisory Committee on Novel Foods (ACNF)에 문의하여 권고를 받을 수 있다(FSANZ, 2024a). ACNF는 해당 식품이 비전통식품 정의에 해당되는지를 판단하고, 비전통식품으로 간주될 경우 안전성 평가의 필요성 여부를 권고한다. 비전통식품에 대한 안전성 평가 필요성을 판단하기 위해서는 잠재적 부작용, 식품의 구성 또는 구조, 제조과정, 유래 원천, 소비패턴 및 수준 및 기타 관련 사항에 대한 정보가 필요하다. ACNF가 식품에 대한 평가를 완료하면 해당 식품이 novel food에 해당하는지에 대한 권고를 제공하고, 이러한 권고내용을 FSANZ 웹 사이트에 공개한다. ACNF의 권고는 법적 조언이 아니라 법적 구속력은 없으며, 해당 식품이 novel food인지 여부에 대한 FSANZ의 조언, 권고 또는 결정 또한 아니다. 이는 문의자가 novel food 신청서를 제출해야 하는지 스스로 결정하는데 도움이 될 수 있도록 하기 위한 절차이고, ACNF로부터 의견을 구하는 것이 법적 의무사항은 아니다. 안전성 평가 필요성 여부에 대한 ACNF의 권고는 시판 전 안전성 평가가 아니라 위해 분석 체계에서 예비 위험성을 확인하는 단계이며, 시판 전 안전성 평가는 FSANZ의 평가 과정의 일부이다.
ACNF에서 novel food 판단을 위한 가이드라인은 다음과 같다(FSANZ, 2024a). 섭취 이력을 판단하는 중요 요소는 사용(섭취) 기간, 사용(섭취집단) 범위, 사용량(섭취 수준) 및 사용 목적으로, 이 네 가지 요소들은 각각 동등하게 간주되지만 특정 요소에서의 부족한 점은 다른 요소에 의해 보완될 수 있다(FSANZ, 2024b).
FSANZ에서는 traditional food, non-traditional food, novel food 및 not novel food 목록을 Novel Foods of the Australia New Zealand Food Standards Code, Standard 1.5.1에 따라 제시하고 있으며, 본 목록에 포함되지 않은 성분들은 요청에 의해 검토 후 포함될 수 있다. FSANZ에서는 전통식품이 아니더라도 안전성에 대한 우려가 없거나, 제3국에서 안전한 섭취이력이 보고된 경우 not novel food로 분류하고 있다. Novel foods는 S25-2 (sale of novel foods) 규정 리스트에 제시되어 있어야만 소매 판매용 식품 또는 식품 성분이 될 수 있다(FSANZ, 2024a).
한편, FSANZ에서는 2007년부터 업체 요구에 따라 특정 상표로 독점 사용 권한(exclusive permission)을 15개월간 허가하고 있다(FSANZ 2007; FSANZ 2024a, c). 그러나, 독점 사용 기간 동안 다른 회사가 다른 회사의 상표로 동일한 novel food에 대해 독점 사용 권한 승인을 신청하는 것을 또한 허용하고 있다. 동일한 식품에 대해 여러 회사가 다른 상표로 독점 사용 권한을 신청할 경우, 후속 신청자의 독점적 사용 기간이 실질적으로 줄어드는 문제가 있다. 따라서, 다수의 회사가 독점 사용 허가를 받은 후 충분한 제품 개발 기간(제품 제형, 디자인, 마케팅 개발 등) 및 식품의 계절적 특성을 고려하여 독점 기간을 18개월 또는 24개월로 연장하는 것을 요청하였으나, Food Regulation Standing Committee가 산업계와 논의 과정에서 도출된 결과를 바탕으로 15개월로 설정되었으므로 이를 유지하는 것으로 결론지었다. 15개월간 동안의 상대적으로 짧은 기간 동안 독점 사용 권한 제한은 독점으로 인한 제품가격 상승이 소비자에게 부정적인 영향을 초래할 수 있고, 15개월 이후 유사 제품을 판매하는 경쟁사와의 시장경쟁을 통해 가격을 낮추기 위함이다.